
Authors: Paul Hofman 1,2, Cécile Badoual 3,4, Fiona Henderson 5
, Léa Berland 1
, Marame Hamila 1
,
Elodie Long-Mira 1,2, Sandra Lassalle 1,2, Hélène Roussel 3,4, Véronique Hofman 1,2
,
Eric Tartour 4,6 and Marius Ilié
1,2,*
1 Laboratory of Clinical and Experimental Pathology, Hospital-Integrated Biobank (BB-0033-00025), Nice Hospital University, FHU OncoAge, Université Côte d’Azur, Nice 06000, France; hofman.p@chu-nice.fr (P.H.); leaberland370@gmail.com (L.B.); hamila.m@chu-nice.fr (M.H.); long-mira.e@chu-nice.fr (E.L.-M.); lassalle.s@chu-nice.fr (S.L.); hofman.v@chu-nice.fr; ilie.m@chu-nice.fr (V.H.)
2 Team 4, Institute for Research on Cancer and Aging, Nice (IRCAN), INSERM U1081/UMR CNRS 7284,
FHU OncoAge, Université Côte d’Azur, Nice 06107, France
3 Department of Pathology, Hôpital Européen Georges Pompidou, APHP, Paris 75015, France; cecile.badoual@aphp.fr (C.B.); helene.roussel@aphp.fr (H.R.)
4
INSERM U970, Université Paris Descartes Sorbonne Paris-Cité, Paris 75015, France; eric.tartour@aphp.fr
5 Department EMEA, Indica Labs, 2469 Corrales Rd Bldg. A-3 Corrales, NM 87048, USA; fhenderson@indicalab.com
6 Department of Immunology, Hôpital Européen Georges Pompidou, Paris 75015, France
* Correspondence: ilie.m@chu-nice.fr; Tel.: +33-0-492-038-263
Abstract
As targeted molecular therapies and immuno-oncology have become pivotal in
the management of patients with lung cancer, the essential requirement for high throughput
analyses and clinical validation of biomarkers has become even more intense, with response
rates maintained in the 20%–30% range. Moreover, the list of treatment alternatives, including
combination therapies, is rapidly evolving. The molecular profiling and specific tumor-associated
immune contexture may be predictive of response or resistance to these therapeutic strategies.
Multiplexed immunohistochemistry is an effective and proficient approach to simultaneously identify
specific proteins or molecular abnormalities, to determine the spatial distribution and activation state
of immune cells, as well as the presence of immunoactive molecular expression. This method is highly
advantageous for investigating immune evasion mechanisms and discovering potential biomarkers
to assess mechanisms of action and to predict response to a given treatment. This review provides
views on the current technological status and evidence for clinical applications of multiplexing and
how it could be applied to optimize clinical management of patients with lung cancer.
1. Introduction
Lung cancer is the leading cause of cancer death among males, and the second most common
among females worldwide [1]. Approximately 80% of newly diagnosed patients with non-small cell
lung cancer (NSCLC) have unresectable locally advanced or metastatic disease [2]. In these patients,
current treatment strategies, across all lines of therapy, include chemotherapy regimens based on
Cancers 2019, 11, 283; doi:10.3390/cancers11030283 www.mdpi.com/journal/cancers Cancers 2019, 11, 283 2 of 22
histology, targeted drugs for patients carrying specific genomic alterations and immunotherapy using
immune checkpoint inhibitors (ICIs), in particular monoclonal antibodies targeting programmed cell
death-1 (PD-1) and programmed cell death ligand-1 (PD-L1) [3–7]. The development of molecularly
targeted therapies, as well as ICIs, has improved outcomes in the metastatic setting for NSCLC patients
who harbor somatically activated oncogenes such as EGFR and BRAFV600, rearranged ALK or ROS1,
or PD-L1 expression ≥50% of tumor cells [3–5]. However, even with these molecular strategies, a large
proportion of patients do not attain prolonged disease control, and the 5-year survival rate does not
exceed 5% [8–10].
Patients with suspected stage IIIB/IV NSCLC require tissue or cytology sampling to confirm the
diagnosis (e.g., adenocarcinoma vs. squamous cell carcinoma vs. other lung histological subtypes),
as this determines eligibility for biomarker testing and further therapeutic strategies [11].
Several immunohistochemical (IHC) markers (e.g., TTF1, p40, INSM1) may be needed to confirm and
subtype lung carcinoma [12,13]. Additional tumor material is required for interrogating predictive
biomarkers, using IHC (e.g., ALK, ROS1, PD-L1), in situ hybridization (ISH; e.g., ALK, ROS1) or
sequencing techniques (e.g., EGFR, BRAF V600E, etc.). Moreover, in the context of precision oncology,
lung cancer patients may be enrolled in ongoing clinical trials (https://clinicaltrials.gov/) and tumor
samples may be used for basic and clinical research studies [14].
For these procedures, sufficient material of high quality is mandatory. In a large number of cases,
the tumor material on which all diagnostic and predictive test must be theoretically be performed
might be sparse, containing only a small number of tumor cells [15]. Small biopsy samples with few
tumor cells might often only allow diagnosis and classification of tumor subtype, and additional tests
may be compromised [11,15].
In the current boost to improve the tailored approach to the clinical management of patients with
NSCLC, pathologists and researchers deal continuously with an unresolved dilemma for exploring a
growing number of protein biomarkers on small-sized tumor samples. In this context, multiplexed
immunohistochemistry (mIHC) has recently emerged as a potent tool for the simultaneous detection
of multiple protein biomarkers on the same tissue section to expand the molecular and immune
profiling of NSCLC, while preserving tumor material. Over the last years, the role of IHC has been
constantly extended to improve diagnosis, and to guide prognosis and treatment of NSCLC patients,
while requiring assessment of an increasing number of protein targets. In addition, multiplying serial
tissue sections to stain for a single marker per slide, can waste small biopsy specimens, entangle the
correlation of section-to-section protein expression, and leave insufficient tumor material for additional
analyses [16]. Multiplexing can be carried out using chromogenic or fluorescent staining methods.
Complex fluorescent multiplexing systems are currently being developed (reviewed in this Special
Focus by Parra et al.) [17]. New approaches compatible with high levels of target multiplexing and
suitable for use on formalin-fixed paraffin-embedded (FFPE) samples have recently demonstrated the
potential to be transferred to the clinical setting [18–22]. For instance, direct simultaneous assessment
by mIHC of both immune and tumor-related pathways and their spatial relationships, in a single tissue
sample, may empower more accurate patient stratification for immunotherapy [23].
Finally, in recent years, mIHC technology has seen rapid advancements in image acquisition
throughput, image resolution and data accuracy, allowing improvements in pathologist performance
by automatically measuring parameters that are hard to achieve reliably by microscope, to extract
comprehensive information on biomarker expression levels, co-localization, and compartmentalization.
The present manuscript reports on mIHC approaches for molecular and immune profiling in
lung cancer.
2. Principles of Multiplexing Staining Methods
2.1. Chromogenic Multiplexed IHC
Technical approaches of brightfield chromogenic mIHC include direct detection of antigens
by primary antibodies from the same or different species that are directly labeled with
different chromogens. Alternatively, an indirect mIHC detection method can be used with two
or more layers of antibodies, allowing for increased amplification of signal [24]. The direct detection
approach has several disadvantages, such as lower sensitivity for low abundance targets, the need
for sizeable quantities of conjugated antibodies, which are usually more expensive, and the risk that
antibody activity could be adversely affected by direct labeling [24]. The indirect approach can be
limited by the number of available host species and the use of same species antibodies, which would
thus require inactivation between successive cycles of immunolabeling [24].
The unwanted cross-reactivity between primary antibodies from different staining cycles is
regarded as the main technical challenge in mIHC. The most frequent solution used to avoid such
reactions is manual microwaving or heating of tissue slides to deactivate the preceding antibody [25,26].
Whereas microwaving is often used in research facilities when dealing with antibodies from the same
host-species, it may not be an optimal method to be adopted in a routine clinical setting. Variable and
heterogeneous results could be obtained by manual processing. Furthermore, microwaving can
increase the damage of the tumor tissue and may remove small biopsies from the slides, especially if
they have already been antigen retrieved by a previous heat-mediated procedure [27].
Another strategy for preventing cross-reactivity is the use of stripping buffers to elute the
primary/secondary antibody complex [27,28]. A number of buffers with different pH, osmolality,
detergent content and denaturing features were evaluated to strip the bound antibody complex from
previous IHC staining cycles, however this produced variable results across studies. Certain buffers
were found to be hazardous, to decolorize H&E stain and/or to reduce nuclear protein staining [27,28].
An alternative, more recent approach named “multiplexed immunohistochemical consecutive
staining on single slide” (MICSSS), was developed for use on FFPE samples by applying
repetitive cycles of immunoperoxidase labeling, image scanning, then chemical stripping of the
chromogenic substrate [20,21]. However, this process can result in a labor-intensive protocol and
a prolonged turnaround time to yield results that are not suitable for a routine clinical setting.
Moreover, multiplexing may be limited due to tissue degradation after successive serial mIHC
cycles [24,29].
More recently, a fully automated mIHC technology using a thermochemical process (heat
deactivation; HD) to deactivate an antibody complex between staining cycles on an automated slide
stainer was first developed for fluorescent detection, and further applied to brightfield chromogenic
detection (Figure 1) [30,31].
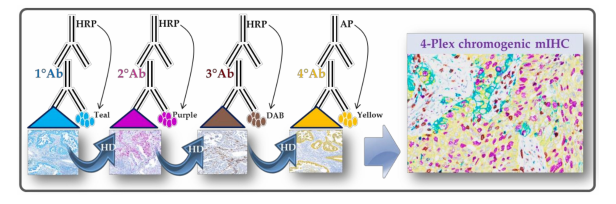 Figure 1. Chromogenic multiplexed immunohistochemistry assay scheme. The assay is using the
sequential application of four unmodified primary antibodies with a specific heat deactivation (HD)
step between staining cycles.
Figure 1. Chromogenic multiplexed immunohistochemistry assay scheme. The assay is using the
sequential application of four unmodified primary antibodies with a specific heat deactivation (HD)
step between staining cycles.This technology allows the use of the first antibody from the same host species, detected by the
anti-species secondary antibody conjugated to horseradish peroxidase (HRP). In the presence of its
substrate, the active HRP, generates in-situ deposition of tyramide within the medium containing
the chromogens. The bound primary antibody/secondary antibody complex is then eluted with a
citrate/acetate buffer. Thus, the deposited chromogen-conjugated tyramide bounds covalently to
the tissue near the first detected antigen. The same procedure is repeated to detect the following
antigens [30,31]. Indeed, the sequential stripping may lead to wastage of the conjugated secondary
antibodies, whereas the chromogen-conjugated tyramine remains stable by binding covalently to
the electron-rich amino acids of detected proteins and by resisting to the elution with the stripping
buffer [30,31]. Importantly, the automation allows for standardization of all critical mIHC steps,
such as HD, reagent application, washing steps, control of temperatures, evaporation and humidity,
while maintaining the integrity of the tissue architecture and the subsequent epitopes [32,33].
To setup a brightfield mIHC assay using sequential detection with unmodified primary antibodies
and chromogenic detection, it is essential to optimize assay conditions on the tissue types of interest
before testing clinical samples [34]. Thus, an optimal mIHC assay needs to assure several staining
performances: (i) equivalent positive/negative signal to single “gold standard” IHC staining, (ii) robust
dynamic proportion of low and high protein quantity, (iii) expected cellular staining topology
(e.g., whole membrane, cytoplasmic, nuclear localization), and (iv) minimal overlap of chromogenic
spectra for co-localized targets [34]. Recent developments have enabled optimal configurations suitable
for testing on clinical samples. For instance, the order of chromogen deposition is determined by the
effect of HD on each epitope, that is, the most HD-affected epitope is incubated first, with the least
affected epitope incubated last.
To offer the best detection sensitivity, other assay parameters must be taken into account such as
the optimal epitope retrieval time to balance the signal/background ratio, and to protect the tissue
architecture by optimizing the incubation time for each primary antibody [30]. Moreover, the number
of antibodies for simultaneous immunolabeling on the same tissue slide has been extended up to
six with the availability of additional chromogens [24,33]. In addition, a major technical challenge
is the risk of insufficient deactivation of the primary antibody complexes, which could determine
cross-reactions and may give false-positive signals. Besides efforts to optimize HD steps during assay
validation, the imaging tools can help to anticipate or to detect potential cross-reactions [35].
2.2. Immunofluorescent Multiplexing
Many newly identified or discovered biomarkers, especially for cancer immunotherapy, are
linked to the tumor microenvironment and need to be analyzed with new methodological tools.
For years, it has become increasingly essential to develop staining and interpretation techniques for
the different cell populations infiltrating or composing a tissue. This is particularly true in oncology.
To date, as previously described, the use of immunohistochemistry can help the visualization of an
antibody-antigen conjugation. It has been showed in the last subsection, that an antibody is conjugated
to an enzyme, like a peroxidase, can catalyze a color-producing reaction. Alternatively, the antibody
can also be tagged with a fluorophore. Nowadays the use of immunofluorescence is far easier due
to technical improvement, like the use of stable fluorophores or the possibility to perform staining in
paraffin embedded slides. Since years, research teams proposed immunoscoring, using single staining
per slide, to identify prognostic factors [36]. However, the tumor microenvironment is too complex to
be summarized by the exploration of a single marker. Chromogenic mIHC is one of the alternatives,
and even if this technique is much easier to be used routinely, it is limited by the use of 4 antibodies
on the same slide. In addition, fluorescence reveals membrane co-localizations (in the membrane
or the nucleus), which is more difficult to obtain with the latter technique. Nevertheless, the use of
multiple antibodies (mixed or used step by step) was restricted to the specificity of the primary and
the risk of false positivity due to cross reactivity between them. Until recently, the single-parametric
Cancers 2019, 11, 283 5 of 22
or even multiparametric (double or triple) staining, revealed by chromomeric or fluorescent staining,
were most often read and interpreted directly by the researchers [37], with a lot of technical constraints.
The microenvironment can now be studied using the multiplex fluorescence technique based on
tyramide coupled to a fluorophore (e.g., Opal®, PerkinElmer, Waltham, MA, USA). This allows the
simultaneous detection of several markers of interest on FFPE tissues. The concept of the technique is
very close the one described above, the chromogenic mIHC assay using sequential application of four
unmodified primary antibodies with a specific HD step between staining cycles. The main advantage
of this technique is the multiplicity of the staining. The technique is based on a conventional fixation
on the epitope of interest. The secondary antibody then binds to the primary antibody followed
by Opal® HRP polymer and one of the Opal®fluorophore adjunction. After deposition of Opal®
reagents, antibodies are stripped after use of a specific microwave to allow subsequent staining of
other antigens. These cycles can be repeated at least seven to nine times. This seven to nine color
multiplex staining technique makes it possible to more precisely characterize different cells and their
interactions with their environment, on the same paraffin slide [38,39]. However, the use of these new
techniques requires the acquisition of specific expertise for in situ multiple staining. Automation of
this different process is now efficient and several autostainers are able to execute most of the steps
previously described.
For the validation of the different panels of multiparametric IHC markers, in particular for the
exploration of the immune system, staining can be performed on tonsil tissue sections as this contains
lympho-epithelial structures (Figure 2). Before any application on a cohort, especially when it concerns
lung sections, the validation of staining on pulmonary tissue sections as a positive control is highly
recommended. In addition, the same positive tissue control could be run on the same slide tested with
mIHC, such as is currently performed for clinical diagnostic IHC.
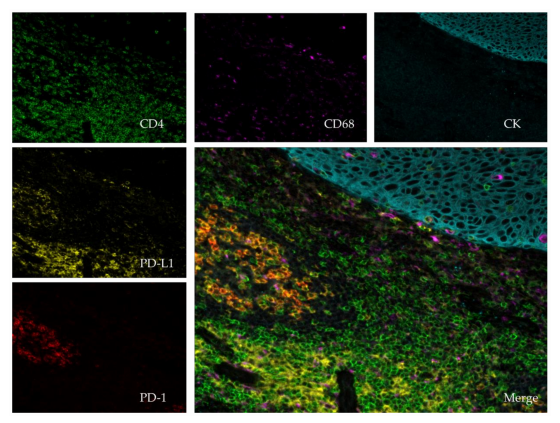
Figure 2. Immunofluorescent multiplexing, image scanned with a spectral scanner (Polaris®;
PerkinElmer, Waltham, MA, USA) using 20× magnification. The tissue is a paraffin embedded
tonsil. The stains are as follows: pan-Cytokeratin (CK, teal), CD4 (green), CD68 (purple), PD-1 (red),
PD-L1 (yellow) and dapi (blue). The central picture compiles the entire staining (merge).
The principle of a multiplex analysis of the tumor microenvironment is the automatic acquisition
of a large surface, or the entire slide, quickly and sustainably. Having a fast acquisition time
(milliseconds for each illuminated spot) is fundamental for fluorescence techniques because it prevents
the "bleaching" which is the progressive extinction of the fluorescent signal after excitation.
3. Clinical and Translational Research Applications: Brief Literature Review and Own Results
Despite the impressive recent achievements in therapeutic strategies for NSCLC treatment, clinical
responses have remained limited to subsets of patients, relapse has occurred in the vast majority of
patients, and only few effective predictive biomarkers have been defined [40]. The development
of more effective predictive biomarkers is needed to optimize patient benefits, minimize the risk of
toxicities, and guide combinatorial approaches. In particular, the emerging picture in immune-oncology
requires a comprehensive understanding of the tumor microenvironment that is the immune landscape
of NSCLC, which results from a complex dynamic cross-talk between the tumor and the immune
system [23,40]. Current efforts on novel biomarker candidates include research on identification
and quantification of different immune cell subsets, their spatial localization and relationships
within tumor areas, the expression of different immune checkpoint markers, tumor mutational
burden, and immune gene signatures [23,40]. Thus, the complete picture will be generated by the
integrative high-dimensional analysis of the tumor and immune profile based on multiple technological
approaches, including mIHC [23].
3.1. Chromogenic Multiplexed Immunohistochemistry
The MICSS technology has demonstrated that high-dimensional characterization of the immune
contexture before and after treatment with ICIs correlates with response to treatment in cancer
patients [20,21]. The immune contexture describes the density, localization, and organization of
the immune cells within solid tumors [41]. By analyzing the composition of complex immune cell
populations, the neutrophil/dendritic cell density score refined the prognostic value of tumors rich in
T-cells and was an independent marker of outcome in NSCLC patients [21].
Another MICSS mIHC platform with computational image processing workflows, including
image cytometry, enabled simultaneous evaluation of three 12-antibody biomarker panels in one
FFPE tissue section, highlighting the impact of in situ monitoring of immune complexity for patient
stratification to improve biomarker discovery and development [20]. The diverse immune complexity
within lymphoid- or myeloid-inflamed tumors as detected by this platform, correlates with clinical
outcomes and tumor sub-classification in head and neck squamous cell carcinoma. In addition,
myeloid-inflamed and T cell exhaustion status correlated with shorter overall survival and the
therapeutic response to vaccination therapy in patients with pancreatic ductal adenocarcinoma [20].
Recently, a chromogenic mIHC method revealed that a high density of tumor-associated
neutrophils (TANs), but not stromal TANs, may have a divergent prognostic effect in NSCLC, negative
in adenocarcinomas, while in squamous cell carcinoma it is a good prognostic factor [42]. Overall,
the in situ high-dimensional assessment of immune cells reveals the potential of mIHC to expand the
immunoscore in NSCLC patients in a clinically relevant manner [43–45].
Interestingly, a recent clinical trial has supported the role for neoadjuvant immunotherapy in
surgically resectable NSCLC, suggesting that the neoadjuvant regimen may lead to early induction of
an adaptive anti-tumor immunity, which could be responsible for preventing distant metastases [6].
While this treatment strategy is still in an early stage of clinical development, there are several pending
questions that are yet to be answered, including whether the major pathologic response could represent
a surrogate end-point for survival and determining the best way to identify upfront patients who
may benefit in this setting [46]. With regard to this, the assessment of candidate biomarkers by
mIHC on tumor biopsies prior to initiation of neoadjuvant treatment as well as on post-treatment
surgical resection samples may be helpful while preserving tumor architecture to assess complete
tumor response. Thus, the mIHC approach could be used to standardize the recently described
“Immune-Related Pathologic Response Criteria” in a clinical setting [47].
Moreover, another open question that remains to be solved is the use of immunotherapy in
special subpopulations, such as elderly patients [48]. Aging is characterized by rebuilding the
immune functions, involving both innate and adaptive immunity [49]. By using a brightfield mIHC
platform, we recently shown that elderly ≥75 years NSCLC patients have less effective anti-tumor
Cancers 2019, 11, 283 7 of 22
immunoreactivity [33]. While further validation in a larger population is required, our findings
suggest that distinct immune pathways may lead to poor outcome in elderly patients with lung
adenocarcinoma [33]. Several previous studies demonstrated that the CD4+/CD8+
ratio may give
more prognostic information than either marker alone in solid tumors [50–52].
As outlined above, mIHC provides a unique sample-sparing analytical tool to characterize limited
clinical tissue samples by multiplexing targets of interest. This method also has the potential to
improve clinical diagnostic accuracy and facilitate histopathological interpretation.
We recently developed in our laboratory (Laboratory of Clinical and Experimental Pathology,
Nice, France) two automated brightfield 4-Plex mIHC assays to comprehensively characterize NSCLC
major histotypes by multiplexing three conventional IHC markers (e.g., TTF1, p40, AE1/AE3)
and three predictive biomarkers (ALK, ROS1, BRAFV600E) cleared by the US Food and Drug
Administration/European Conformity-In Vitro Diagnostic (FDA/CE-IVD) [22]. Some pathology
laboratories use chromogenic mIHC on FFPE samples but stain for no more than two markers per
tissue slide [45]. The two assays demonstrated no antigenicity loss, steric interference or increased
cross-reactivity, providing an analytical tool that can be integrated in a routine clinical workflow [22].
In addition, there are some concerns on the extent to which a multi-color background with color overlap
on whole-slide samples could influence the visual interpretation of critical biomarkers. In particular,
the PD-L1 expression can be heterogeneous and variably expressed in either tumor or immune
cells [53]. By excluding the PD-L1 expressing cells that are unstained with keratin and TTF1 as per
tumor-infiltrating immune cells expressing PD-L1, the chromogenic mIHC assay made the visual
interpretation straightforward and less ambiguous (Figure 3).
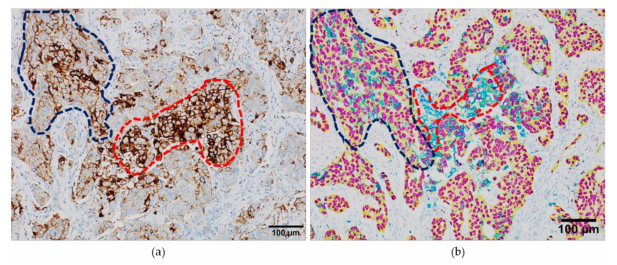
Figure 3. Interpretation of the programmed death-ligand 1 (PD-L1) staining in serial whole-tissue
formalin-fixed paraffin embedded samples from a lung adenocarcinoma case. (a) PD-L1 expression
revealed by conventional immunoperoxidase staining; (b) PD-L1 expression revealed by chromogenic
multiplexed immunohistochemistry, with the anti-TTF1 antibody colored in purple, anti-AE1/AE3 in
yellow and anti-PD-L1 SP263 in teal. Blue dotted line: tumor area; red dotted line, immune cells.
As the restricted tissue size is a major issue for the management of the vast majority of solid tumors,
and individual antibodies rarely demonstrate 100% specificity in the determination of malignancy
or cell lineage, a chromogenic mIHC approach with specific multiple protein markers can provide
valuable diagnostic information and has the potential to enhance the clinical significance of histological
subtyping by delivering substantial prognostic information with therapeutic consequences [54,55].
3.2. Immunofluorescent Multiplexing
3.2.1. Localization of Immune Cells and Their Relationships with Immunosuppressive Markers in the
Tumor Microenvironment
The multiplex immunofluorescence techniques better distinguish the stromal and the tumor
compartment and thus have allowed for a more detailed description of the topography of immune
Cancers 2019, 11, 283 8 of 22
cells in cancer. Cruz et al. found that T lymphocytes were predominantly concentered in stromal
compartment instead of the epithelial compartment in NSCLC [56]. Based on a quantitative
immunofluorescence study, a comparative analysis of the expression of immunosuppressive molecules
(e.g., PD-L1, IDO-1, B7H4) with the infiltration of intratumoral cells in lung cancer showed that PD-L1
and IDO-1 were consistently associated with prominent B- and T-cell infiltrates, but B7-H4 was not [57].
This could be explained by the role of IFNγ produced by immune cells in regulating PD-L1 and IDO-1
in the tumor microenvironment [58].
3.2.2. Novel Prognostic Composite Biomarker based on Fluorescence in Situ Multiplexing
One of the first clinical studies based on fluorescent digital pathology was the work of
Schalper et al., who reported that the infiltration of intratumoral CD3+ and CD8+T cells was associated
with a better overall survival in lung cancer patients [59]. For the CD8+T cell infiltration, this prognostic
impact was independent from age, tumor size, histology and stage in multivariate analyses [59].
This technology also allows us to better define the prognostic value of immune cells depending on
their localization in the tumor microenvironment. For example, after neoadjuvant chemotherapy, high
levels of epithelial but not stromal CD4+CD3+T lymphocytes correlated with better survival in patients
with NSCLC [60].
A more complex cell phenotype could also be better characterized with this multiparametric analysis.
A novel subpopulation of CD8+T cells called resident memory T cells appear to play a major role in
immunosurveillance, as they localize in close contact with epithelial tumor cells [61]. They are defined by a
composite phenotype including various biomarkers such as CD103, CD49a, CD69 (Figure 4).

Figure 4. Infiltration of resident memory T cells (CD103+CD8+T cells) in human lung cancer.
Frozen tissue sections derived from lung adenocarcinoma patients were stained by immunofluorescence
with antibodies directed against human (a) CD8 (green), and (b) CD103 (red). (c) The co-localization
of CD8 and CD103 markers can be detected by merging the mono-stained pictures. The arrows
indicate double positive cells. Staining with isotype controls was included for each experiment (20×
magnification).
We previously demonstrated that high levels of intratumoral infiltration with a resident memory
CD8+T cells are associated with a better clinical outcome of NSCLC patients, both in univariate and
multivariate analyses [62]. These were a more powerful prognostic marker than the infiltration of total
CD8+T cells. These data were then confirmed by various clinical studies [63,64].
This technique also allows us to focus beyond just one cell type, and to integrate the relationships
that exist between immune cells in the various compartments of tumors and the relative impact of these
cellular relationships on the future of patients. For example, a high effector CD8+T cell/regulatory
T cell ratio in the tumor nest is correlated with a better overall survival than when each cell measured
independently [65].
3.2.3. Fluorescence Multiplexing Technique to Predict Clinical Response to Immunotherapy
Various parameters such as PD-L1, the expression of PD-1 and the intratumoral infiltration of
CD8+T cells are considered, especially when combined together, as potential predictive biomarkers of
clinical response to immunotherapy [66]. Parra et al., observed higher levels of PD-L1 expression on
Cancers 2019, 11, 283 9 of 22
tumor cells and an increase in the infiltration of T cells and PD-1+T cells in the tumor microenvironment
of NSCLC after neoadjuvant chemotherapy [60]. These findings confirm studies in other cancers
reporting that neoadjuvant chemotherapy, whatever the regimen, makes the tumor microenvironment
more permissive to immunotherapy [67,68]. These results suggest that it would be worthwhile to
combine chemotherapy and immunotherapy before surgical resection of locally advanced lung cancer.
Using a quantitative multiplex immunofluorescence technique, we reported that EGFR-mutated
NSCLC weakly expressed PD-L1 and was not infiltrated by CD8+T cells suggesting that it would not
be prone to respond to immunotherapy [69]. This hypothesis was then clinically confirmed in various
clinical trials [70]. Interestingly, we found that a subpopulation of NSCLC displaying chromosomal
rearrangement of the ALK gene expressed significant levels of PD-L1 on their tumor cells and were
infiltrated by PD-1+CD8+T cells [69]. However, other studies showed that concurrent CD8+T cells and
high PD-L1 expression on tumor cells tend to be rare in ALK positive NSCLCs [71,72]. Clinical trials
did not confirm the sensitivity of this cancer subtype to the blockade of PD-1/PD-L1 axis [71].
This may suggest that other resistance mechanisms occur in this population such as the possible
co-expression of inhibitory receptors on T cells or the infiltration of immunosuppressive cells [73,74].
Finally, an increase of T cells with a quiescent phenotype defined by a low proliferation and activation
status (Ki67 and Granzyme negative) correlated with better overall survival in NSCLC patients
treated by anti-PD-1/PD-L1 [75]. Interestingly, in NSCLC patients not treated by immunotherapy, this
population of “dormant” T cells did not correlate with a better clinical outcome, supporting the fact
that these cells could represent a true predictive biomarker of response to immunotherapy and not a
prognostic marker [75].
4. Image Analysis of Multiplexed Staining
Until recently, pathologic analysis of the IHC signal remained a subjective and time-consuming
procedure, wherein the staining intensity, localization and amount had to be manually assessed.
Therefore, despite development of practical scoring systems, such as the H-score, the scoring decision
is still directly influenced by visual bias [76,77]. Nowadays, with the advent of precision digital
immune-oncology, pathologists face a technological transition phase. The convergence of tissue-based
mIHC along with automated computer-aided imaging technologies has the potential to make complex
information more accessible in routine clinical workflows, improving prognostic and predictive
patient stratification [78]. Image analysis and artificial intelligence tools and fields of application in
immune-oncology have been outlined in a recent review by Koelzer et al. [78].
The improvement in digital imaging processing systems has opened new doors towards an
unbiased, unsupervised, and automatic IHC image analysis by measurement of optical density,
which is proportional to the expression extent of specific antigens [77]. Furthermore, application of an
automated scoring method for mIHC signals might help pathologists in quantitative comparisons and
produce a more accurate characterization of the tumor microenvironment. The mIHC digital image
must have the correct stains unmixed into their constituent chromogens for each individual biomarker.
Moreover, in order to obtain accurate identification, segmentation and profiling of tumor and immune
cells, the mIHC image analysis has to assure the same quantity of chromogen in the color mixture [35].
Several technologies have been developed to decompose each pixel into a collection of constituent
signals and the fractions from each of them, in order to convert the whole image into analyte-specific
image channels [79]. However, the maximum number of stains that can be unmixed was limited
to three, as the linear system had insufficient equations for cases of more than three stains [35].
Alternatively, a novel multi-spectral image deconvolution algorithm has been developed to handle
more than three colors and to maintain the biological properties of the protein markers [35].
An increasing number of automated digital pathology systems are being used to analyze
information from mIHC technology, such as HALO (Indica Labs, London, UK) [80] for up
to five colors, Vectra/inForm (PerkinElmer, Waltham, MA, USA) for up to three colors [81],
the “Aperio Color Deconvolution Algorithm” or SlidePath (Leica Biosystems, Wetzlar, Germany) for
Cancers 2019, 11, 283 10 of 22
up to three colors [82], BLISS workstation (Bacus Laboratories, Lombard, IL, USA) for up to four colors
but restricted to one region-of-interest (ROI), Tissue Studio® 4.0 (Definiens, Munich, Germany) for up
to two colors [83], the “Automated Cellular Imaging System” (ACIS III, Dako, Glostrup, Denmark),
and Mirax HistoQuant (3DHistech, Budapest, Hungary) [84].
In our own experience we have used HALO, which is an automated quantitative digital pathology
platform, compatible with all major microscope/slide scanners and non-proprietary tiff/jpeg formats
and allowing for whole-slide and field-of-view analyses. Modules used for mIHC analysis include
mIHC (brightfield mIHC), a tissue classifier module for tissue differentiation (e.g., tumor vs. stroma),
and a spatial analysis module for interrogating spatial distributions of cell populations within the
same, or serial tissue sections. Occasionally, it is critical to separate out the tumor and stroma into
two classes, in order to determine the percentage of tumor cells positive for x, versus the percentage of
stromal cells positive for x. Manually annotating these regions is extremely laborious and therefore
automatic detection of these two regions is required for high-throughput analysis. HALO uses two
different machine learning classifiers for automatic tissue detection: the random forest classifier and
HALO-AI. The random forest classifier uses the random forest algorithm to assign pixels to a certain
class based on color and texture. A random forest classifier is very quick to create and is effective in
applications such as differentiating between tumor and stroma as shown in Figure 5. The Serial Section
module also allows one to create a classifier on one stain (e.g., an H&E image), and then superimpose
the classification onto a registered serial section. Therefore, there is no need to have a tumor marker on
each serial section to achieve tumor/stroma separation.
The random forest classifier is quick and easy to set-up but will often suffer when presented
with multiple variable tissue staining; such is often true for large clinical cohorts. In such situations
HALO-AI, a deep learning classifier can be used. HALO-AI is a convolutional neural network for
pattern recognition within a tissue section. Whilst a pathologist’s input is increased relative to random
forest, the training results in a highly robust classifier that can be used across large cohorts. HALO-AI
can even be trained to recognize different tissue classes across different stains. The probability map
and conversion to annotation features can also be used in HALO-AI.
Once the selected classifier has been created and saved, it can then be used in the mIHC analysis
in HALO. In brightfield, the mIHC module allows the pathologist to detect up to 5 stains, including an
exclusion stain, in any cell compartment (nucleus, cytoplasm, membrane). The exclusion stain option can
be used to exclude tar within lung tissue. An example of a mIHC analysis in HALO is shown in Figure 6.
Prior to running the mIHC analysis, pathologists can define specific phenotypes such as active T
cells (e.g., dual-positive cells for brown and purple stains will be identified as dual positive for CD8
and Ki67; Figure 6).

Figure 5. Tissue classification using the random forest classifier in non-small cell lung cancer tissue
(20× magnification). (a) The multiplexed immunohistochemistry (mIHC) image was scanned with a
Nanozoomer 2.0-HT Scanner (Hamamatsu photonics, Hamamatsu, Japan). The stains are as follows:
Pan-cytokeratin (yellow), CD8 (brown), Ki67 (purple), PD-L1 (teal) and hematoxylin (dark purple).
(b) The random forest classifier in HALO was used to separate the image into three classes: tumor,
stroma and microscope glass slide. The classifier mask is shown overlaying the mIHC image where
classified tumor regions are shown in yellow, stroma regions in purple, and the microscope glass slide
in pale pink. (c) The probability threshold used by the random forest to detect tumor regions was
increased to 70%. A heatmap is displayed where the red regions represent areas most likely to be tumor
regions, and the green regions that are less likely. No mask will appear in areas where pixels have
below 70% probability of being in the tumor class. (d) The classifier to annotations option was used
whereby regions can automatically be annotated from the classification mask; only the tumor has been
annotated (shown in yellow).
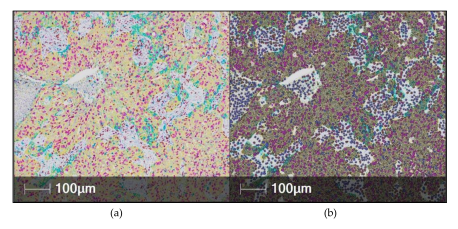
Figure 6. Automated digital analysis of multiplexed immunohistochemistry (mIHC) using the HALO
software in non-small cell lung cancer tissue. (a) The mIHC image was scanned with a Nanozoomer
2.0-HT Scanner (Hamamatsu photonics, Hamamatsu, Japan) using 20x magnification. The stains
are as follows: Pan-cytokeratin (yellow), CD8 (brown), Ki67 (purple), PD-L1 (teal) and hematoxylin
(dark purple). (b) The HALO mark-up image shows colors similar to the original stain color and in
the same cell compartment (nucleus/cytoplasm/membrane as the stain is found. The user can select
different colors to be used in the mark-up image if they wish.
When this is run in conjunction with the pre-made classifier, information about the number of cells
with a specific phenotype in both the tumor and the stroma can be obtained. Additionally, HALO’s
interactive cell-by-cell data table allows easy localization of the phenotyped cells on the image. In the
example analysis in Figure 7, outputs will include those for the entire image, those specific to the tumor
and those specific to the stroma. Other outputs include the number of cells positive for each stain in
each compartment, number of cells with different stain co-localizations, the average optical density
values for each stain in each compartment, cell/nucleus/cytoplasm/membrane area, and tissue areas
in square microns.
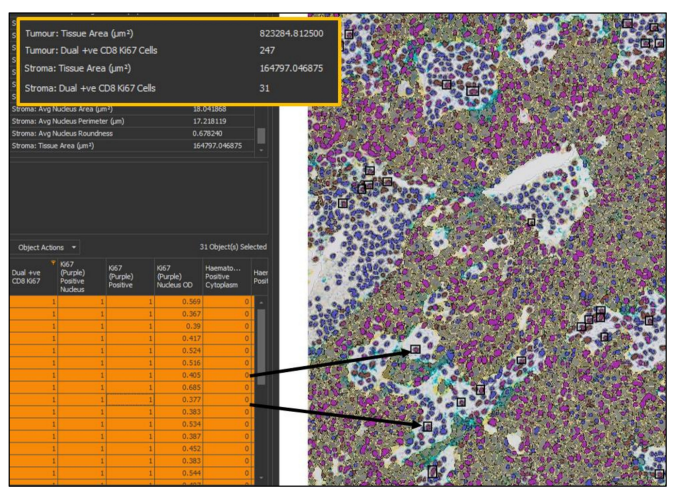
Figure 7. Results of a multiplexed immunohistochemistry (mIHC) analysis in HALO. The top left table
provides the summary results from the analysis; important outputs in this analysis are the density
of cells co-expressing both CD8 and Ki67 in the tumor and stroma, and so the data relating to this
has been highlighted. The bottom left table is HALO’s interactive cell-by-cell data table, which can
be mined to find specific cell types. Here, only cells that are positive for CD8 and Ki67 and are in
the stroma have been selected. HALO will find the cells selected in the image viewer (right, 20×
magnification) by putting a black box around each cell.
After running a mIHC analysis in HALO, the pathologist then has the option to generate
spatial information using the spatial analysis module. As outlined above, spatial information is
becoming increasingly important in cancer research, prominently in the immune-oncology field [36,85].
Three different types of spatial analysis can be performed in HALO: nearest neighbor, proximity
analysis and invasive margin analysis. Nearest neighbor outputs will calculate the average distance of
two cell populations based on their nearest neighbors. Proximity analysis allows you to calculate the
number of cells of one phenotype (e.g., CD8+ cytotoxic T lymphocytes) within a defined distance of
another cell type. Lastly, the invasive margin analysis allows you to count the number of cells within a
user defined distance of the invasive margin.
Similarly, the HALO image analysis software was recently used to demonstrate the divergent
state of exhaustion of the PD-1 receptor in T cells with impaired effector cytokine production,
while producing CXCL13, which mediates immune cell recruitment to tertiary lymphoid structures [80].
Cancers 2019, 11, 283 13 of 22
Importantly, the presence of PD-1high cells was strongly predictive for both response and survival in a
cohort of NSCLC patients treated with a PD-1 blocking agent [80].
In the immunofluorescence multiplexing field, the use of scanners (fluorescent or spectral)
represents a major technological advance by enabling the utilization of multiple and sometimes unstable
fluorochromes (e.g., phycoerythrins) and thus more than 7 different antibodies on the same slide.
For example, Vectra® systems or Polaris®(PerkinElmer) allow the capture of information by spectral
resolution in the visible and in the near infrared band (bandwidth between 420 and 900 nm). Vectra®
or Polaris® allows extremely precise quantitative (cell-by-cell) management of the markings of different
tissue samples, in brightfield or fluorescence detection. Detection and phenotypic characterization of
cells in tissues, combined with bioinformatic image analysis is possible thanks to the InForm® software
(PerkinElmer). This software allows automatic analysis of parameters that cannot be accurately discerned
by the human eye (cell forms, multiple molecule networks, vascular network).
Franchising of autofluorescence by the "Autofluorescence Reduction Technology” (ART™,
PerkinElmer) technique is possible with the Inform® software (PerkinElmer). Of course,
the technologies developed for a specific type of cancer are subsequently transposable to the majority
of other tumor proliferations or inflammatory diseases. Finally, virtual slides can be analyzed
automatically (cell counting, surface measurements, etc.) using dedicated image analysis software
(Figure 8). In particular, some software enables the quantification of weakly expressing and overlapping
biomarkers within cells and cellular compartments.

Figure 8. Automated digital analysis of fluorescent multiplexing using Inform software in tonsil tissue
(20× magnification). (a) Multiparametric fluorescent staining Pan-Cytokeratin (turquoise), CD4 (green),
CD68 (purple), PD-1 (red), PD-L1 (yellow) and dapi (blue). (b) Tissue segmentation: identification and
recognition of tumor areas (red) or stroma (green). (c) Individual cells identification and segmentation,
with nuclear, membranous and cytoplasmic segmentation. (d) Phenotyping: identification of the cells
on the slide, with their phenotypes, among all the cells present in the image, or among the cells stained.
These new approaches allow us to explore cellular interactions to find biomarkers in a
non-supervised manner. The education of the software remains long and tedious, with a phase
of learning or "teaching". However, an approach without a priori knowledge can also be developed
in parallel. Several companies developed such software (e.g., Definiens AG, Munich, Germany;
TRIBVN Healthcare, Châtillon, France; Owkin, Paris, France; Imstar, Paris, France; Indica Labs;
PerkinElmer). These software systems are becoming more and more efficient, and they can differentiate
anatomical structures, such as glands [86], but the recognition of cell units is more delicate. The results
obtained in the context of cross-sectional research studies are, however, very impressive and we must
expect a change in diagnostic habits with the implementation of deep learning [87].
Finally, an important issue for mIHC digital analysis and relevant data extraction is the calibration
of the signal acquisition technology and the control of variations caused by the different staining
techniques when several batches are required to analyze large clinical series (e.g., for biomarker validation).
These controls are also necessary for the valid comparison of different series or studies and ultimately for
clinical application [88].
5. Advantages and Current Limitations of Multiplexed Immunohistochemistry
Recently developed multiplexing platforms exhibit compelling advantages. The major advantage
of mIHC, which may also warrant its implementation in the routine clinical workflow, is related
to maximal data harvesting per tissue section, improvement in the quality and detail of pathology
analysis and efficient tissue utilization, which is crucial when the availability of sample is limited [89].
Approaches like mIHC enable pathologists to gather a wealth of data from a limited amount of tissue.
This is especially promising for NSCLC patients whose tumors are in a difficult-to-access location,
where only a small needle or cytology sample can be obtained. It also enables more research to be
conducted with less material than is often required [89]. Unlike other multiplex approaches, such
as next generation sequencing or mass spectrometry, mIHC gives an edge to analyze co-expression
and to quantify single-cell expression with the spatial relationships of many targets while preserving
tissue integrity. Several studies have shown that the proximity of certain immune cells within a tumor
microenvironment correlates with patient outcome [41,85,90].
Recently developed strategies in the field of brightfield chromogenic mIHC have enabled
automation of mIHC assays through the use of commercially available primary antibodies with
their respective anti-species secondary antibody to ensure staining reliability and reproducibility,
toward the clinical application [22]. Moreover, conventional brightfield microscopes and scanners can
accommodate image acquisition of the stained slides [78].
However, multiple pre-analytical and analytical challenges arise when using chromogens for
high-level mIHC analysis. The limited number of available chromogens, compared to highly
multiplexed fluorescent assays, limits the degree of flexibility for biomarker research. As chromogenic
mIHC is technically similar, in some ways, to conventional IHC it is subjected to the same critical
hurdles [91]. The lack of standardization due to pre-analytic variables, including fixation time, type
of fixative, dehydration, clearing, paraffin impregnation, and drying and storage of the slides, still
represents a major potential challenge [92]. Similarly, poorly characterized or cross-reactive antibodies
will give non-reproducible results [93]. For instance, despite numerous efforts to standardize the IHC
markers used in breast cancer (ER/PR/HER2), they still demonstrate significant inter-laboratory and
intra-laboratory variability [94]. If such issues cannot be overreached for these “conventional” IHC
biomarkers, the multiplexing of several markers will need sufficient robustness prior to a clinical use.
As for the clinical single IHC assays, a positive tissue control previously validated and characterized
should be run on each same slide tested with mIHC. This would allow “real-time” validation of the
multiplexed staining along with the quality control of data generated by the mIHC assay.
As tumors frequently harbor significant cellular and spatial heterogeneity (e.g., stroma,
tumor-stroma interface, intratumoral), in particular for immune markers such as PD-L1 or CD8
infiltrates [95], it is essential to perform high-resolution multiplexed analysis across whole tumor
Cancers 2019, 11, 283 15 of 22
sections. It has been demonstrate that the analysis of small ROIs generates significant variation and
errors in the assessment of tumor and immune markers in cancer [96,97]. Hence, there is a need
for integrated mIHC systems enabling high-degree of multiplexing coupled with digital analysis for
high-resolution analyses on whole tumor slides [98].
Moreover, mIHC is the only technology enabling quantitative information on multiple
distinct subtypes of tumor-infiltrating immune cells within a preserved tissue architecture, hence
allowing the analysis of the topology and proximity between specific cell populations [99].
Ultimately, the quantitative spatial profiling of key tumor-immune pathways could improve the
stratification of cancer patients for immunotherapy [100]. In addition, the explosion of potentially
important or actionable biomarkers poses both cost and selection challenges. The increase in
the number of developed chromogens could make this challenge somewhat easier to handle [16].
However, the current cost of the primary antibodies or different chromogens and the instrumentation
requirements are still high. More than four antibodies can be sequentially incubated on autostainers,
reducing the difficulty, delay and therefore cost to perform the mIHC analysis in a clinical setting,
although, as noted above, pre-analytical variability and antigen retrieval methods will first need to be
critically evaluated. Moreover, evaluation of multiple targets per tissue slide will require digital image
viewing with analysis tools for computer-assisted interpretation that are yet to be readily integrated in
the clinical workflow [78]. For a wide clinical implementation and pathologists’ acceptance, regulatory
and reimbursement rules should be planned in the near future. Nevertheless, the extraordinary value
of such a technological approach to improve pathology interpretation and to yield new insights into
understanding cancer phenotypes with direct clinical impacts warrants further effort.
The different considerations presented above could be declined for the fluorescent mIHC.
The specificity of the staining has been improved with the use of tyramide techniques allowing
simultaneous staining with 7 to 9 colors in a same slide. The different technical implementations
described in this article have to reinforce the efforts made to increase the knowledge about
microenvironment. Fluorescent staining keeps an advantage in research for the observation of very
rare events, rare cells, co-localization and still allows a better study of the different cell compartments.
Nevertheless, this technique is still difficult to be used in routine; the signal reproducibility is difficult
to be obtained, even with an automation of the staining.
Several alternative multiplexed technologies for a use on FFPE samples have recently been
developed (e.g. multiplexed ion beam imaging-MIBI, IONpath, Inc., Menlo Park, CA, USA; imaging
mass cytometry, Fluidigm, South San Francisco, CA, USA; digital spatial profiling technology,
NanoString Technologies, Inc., Seattle, WA, USA; InSituPlex, Ultivue, Cambridge MA, USA)
demonstrating a high degree of multiplexing, and could be complementary to mIHC approaches
described herein [89,101–103].
6. Conclusions
Technological advances in mIHC and the introduction of automated slide scanners has allowed
for huge amounts of data to be generated in a single experiment. Combining this with automated
digital analysis means the data can be analyzed in a quantitative and efficient manner, producing a
high-throughput workflow for molecular and immune profiling with the promise of discovering novel
biomarkers and improving clinical management of patients with NSCLC.
---------------------------
PDF Version: cancers-11-00283.pdf
Author Contributions:
Conceptualization, P.H., C.B. and M.I.; methodology, P.H., E.T., F.H., C.B., and M.I.;
software, E.T., F.H., C.B., and M.I.; validation, P.H., E.T., F.H., C.B., and M.I.; formal analysis, P.H., E.T., F.H., C.B.,
and M.I.; investigation, P.H., E.T., F.H., L.B., E.L-M., S.L., H.R., V.H., C.B., and M.I.; resources, P.H., E.T., F.H., L.B.,
E.L-M., S.L., H.R., V.H., C.B., and M.I.; data curation, P.H., E.T., F.H., L.B., E.L-M., S.L., H.R., V.H., C.B., and M.I.;
writing—original draft preparation, P.H., E.T., F.H., C.B., and M.I.; writing—review and editing, P.H., E.T., F.H.,
L.B., E.L-M., S.L., H.R., V.H., C.B., and M.I.; visualization, P.H., E.T., F.H., C.B., and M.I.; supervision, P.H., E.T.,
F.H., C.B., and M.I.; project administration, P.H., E.T., F.H., C.B., and M.I.; funding acquisition, P.H., E.T., F.H., C.B.,
and M.I.
Cancers 2019, 11, 283 16 of 22
Funding: This research was funded in part by the Initiative of excellence IDEX - UCAJEDI, Université Côte d’Azur;
the Cancéropole PACA; the French Government (Agence Nationale de Recherche, ANR) through the ‘Investments
for the Future’ LABEX SIGNALIFE [ANR-11-LABX-0028-01]; CARPEM (SIRIC); Labex Immuno-Oncology, INCA
(2016-PL-Bio); the “Fondation ARC pour la Recherche sur le Cancer” (ARC SL220110603478); the CANC’AIR
Genexposomic project; the “Ligue Départementale 06 de Lutte contre le Cancer”, the “Conseil Départemental
des Alpes-Maritimes”; the “Région Sud-Provence Alpes-Côte d’Azur”, France. The funders had no role in the
design of the study; in the collection, analyses, and interpretation of data; in the writing of the manuscript, or in
the decision to publish the results.
Acknowledgments: To Julien Fayada and Marion Mandavit for their technical support.
Conflicts of Interest: P.H. has received travel grants and honoraria from AstraZeneca, Roche, Bristol-Myers
Squibb, Qiagen, ThermoFisher, Pfizer, and Merck & Co. F.H. is an employee of Indica Labs. C.B. has received grant
travel and/or honoraria from AstraZeneca, Roche, Bristol-Myers Squibb, and Merck & Co. E.T. has received travel
grants and honoraria from AstraZeneca, Bristol-Myers Squibb. M.I. has received travel grants and honoraria from
AstraZeneca, Merck & Co., Roche, Boehringer-Ingelheim and Bristol-Myers Squibb. There are no other competing
financial interests or other conflicts.
References
1. Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018:
GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer
J. Clin. 2018, 68, 394–424. [CrossRef] [PubMed]
2. Scagliotti, G.V.; Bironzo, P.; Vansteenkiste, J.F. Addressing the unmet need in lung cancer: The potential of
immuno-oncology. Cancer Treat. Rev. 2015, 41, 465–475. [CrossRef] [PubMed]
3. Lee, C.K.; Brown, C.; Gralla, R.J.; Hirsh, V.; Thongprasert, S.; Tsai, C.M.; Tan, E.H.; Ho, J.C.; Chu da, T.;
Zaatar, A.; et al. Impact of EGFR inhibitor in non-small cell lung cancer on progression-free and overall
survival: A meta-analysis. J. Natl. Cancer Inst. 2013, 105, 595–605. [CrossRef] [PubMed]
4. Shaw, A.T.; Engelman, J.A. ALK in lung cancer: Past, present, and future. J. Clin. Oncol. 2013, 31, 1105–1111.
[CrossRef] [PubMed]
5. Schiller, J.H.; Harrington, D.; Belani, C.P.; Langer, C.; Sandler, A.; Krook, J.; Zhu, J.; Johnson, D.H. Comparison
of four chemotherapy regimens for advanced non-small-cell lung cancer. N. Engl. J. Med. 2002, 346, 92–98.
[CrossRef] [PubMed]
6. Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.;
Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018,
378, 1976–1986. [CrossRef] [PubMed]
7. Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.;
Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung
Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [CrossRef] [PubMed]
8. Reck, M.; Popat, S.; Reinmuth, N.; De Ruysscher, D.; Kerr, K.M.; Peters, S.; Group, E.G.W. Metastatic
non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and
follow-up. Ann. Oncol. 2014, 25, iii27–iii39. [CrossRef] [PubMed]
9. Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355.
[CrossRef] [PubMed]
10. Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.;
Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis,
treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [CrossRef] [PubMed]
11. Dietel, M.; Bubendorf, L.; Dingemans, A.M.; Dooms, C.; Elmberger, G.; Garcia, R.C.; Kerr, K.M.; Lim, E.;
Lopez-Rios, F.; Thunnissen, E.; et al. Diagnostic procedures for non-small-cell lung cancer (NSCLC):
Recommendations of the European Expert Group. Thorax 2016, 71, 177–184. [CrossRef] [PubMed]
12. Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.;
Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of
Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260.
[CrossRef] [PubMed]
13. Inamura, K. Update on Immunohistochemistry for the Diagnosis of Lung Cancer. Cancers 2018, 10, 72.
[CrossRef] [PubMed]
Cancers 2019, 11, 283 17 of 22
14. Giraldo, N.A.; Peske, J.D.; Sautes-Fridman, C.; Fridman, W.H. Integrating histopathology, immune
biomarkers, and molecular subgroups in solid cancer: The next step in precision oncology. Virchows Arch.
2019. [CrossRef] [PubMed]
15. Coghlin, C.L.; Smith, L.J.; Bakar, S.; Stewart, K.N.; Devereux, G.S.; Nicolson, M.C.; Kerr, K.M.
Quantitative analysis of tumor in bronchial biopsy specimens. J. Thorac. Oncol. 2010, 5, 448–452. [CrossRef]
[PubMed]
16. Levenson, R.M.; Borowsky, A.D.; Angelo, M. Immunohistochemistry and mass spectrometry for highly
multiplexed cellular molecular imaging. Lab. Investig. 2015, 95, 397–405. [CrossRef] [PubMed]
17. Parra, E.R.; Francisco-Cruz, A.; Wistuba, I.I. State-of-the-Art of Profiling Immune Contexture in the Era of
Multiplexed Staining and Digital Analysis to Study Paraffin Tumor Tissues. Cancers 2019, 11. [CrossRef]
[PubMed]
18. Blom, S.; Paavolainen, L.; Bychkov, D.; Turkki, R.; Maki-Teeri, P.; Hemmes, A.; Valimaki, K.; Lundin, J.;
Kallioniemi, O.; Pellinen, T. Systems pathology by multiplexed immunohistochemistry and whole-slide
digital image analysis. Sci. Rep. 2017, 7, 15580. [CrossRef] [PubMed]
19. Gorris, M.A.J.; Halilovic, A.; Rabold, K.; van Duffelen, A.; Wickramasinghe, I.N.; Verweij, D.; Wortel, I.M.N.;
Textor, J.C.; de Vries, I.J.M.; Figdor, C.G. Eight-Color Multiplex Immunohistochemistry for Simultaneous
Detection of Multiple Immune Checkpoint Molecules within the Tumor Microenvironment. J. Immunol. 2018,
200, 347–354. [CrossRef] [PubMed]
20. Tsujikawa, T.; Kumar, S.; Borkar, R.N.; Azimi, V.; Thibault, G.; Chang, Y.H.; Balter, A.; Kawashima, R.;
Choe, G.; Sauer, D.; et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed
Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 2017, 19, 203–217. [CrossRef]
[PubMed]
21. Remark, R.; Merghoub, T.; Grabe, N.; Litjens, G.; Damotte, D.; Wolchok, J.D.; Merad, M.; Gnjatic, S. In-depth
tissue profiling using multiplexed immunohistochemical consecutive staining on single slide. Sci. Immunol.
2016, 1, aaf6925. [CrossRef] [PubMed]
22. Ilie, M.; Beaulande, M.; Hamila, M.; Erb, G.; Hofman, V.; Hofman, P. Automated chromogenic multiplexed
immunohistochemistry assay for diagnosis and predictive biomarker testing in non-small cell lung cancer.
Lung Cancer 2018, 124, 90–94. [CrossRef] [PubMed]
23. Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint
inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [CrossRef]
24. Stack, E.C.; Wang, C.; Roman, K.A.; Hoyt, C.C. Multiplexed immunohistochemistry, imaging,
and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and
multiplex analysis. Methods 2014, 70, 46–58. [CrossRef] [PubMed]
25. Osman, T.A.; Oijordsbakken, G.; Costea, D.E.; Johannessen, A.C. Successful triple immunoenzymatic method
employing primary antibodies from same species and same immunoglobulin subclass. Eur. J. Histochem.
2013, 57, e22. [CrossRef] [PubMed]
26. Feng, Z.; Puri, S.; Moudgil, T.; Wood, W.; Hoyt, C.C.; Wang, C.; Urba, W.J.; Curti, B.D.; Bifulco, C.B.; Fox, B.A.
Multispectral imaging of formalin-fixed tissue predicts ability to generate tumor-infiltrating lymphocytes
from melanoma. J. Immunother. Cancer 2015, 3, 47. [CrossRef] [PubMed]
27. Pirici, D.; Mogoanta, L.; Kumar-Singh, S.; Pirici, I.; Margaritescu, C.; Simionescu, C.; Stanescu, R.
Antibody elution method for multiple immunohistochemistry on primary antibodies raised in the same
species and of the same subtype. J. Histochem. Cytochem. 2009, 57, 567–575. [CrossRef] [PubMed]
28. Gendusa, R.; Scalia, C.R.; Buscone, S.; Cattoretti, G. Elution of High-affinity (>10–9 KD) Antibodies
from Tissue Sections: Clues to the Molecular Mechanism and Use in Sequential Immunostaining.
J. Histochem. Cytochem. 2014, 62, 519–531. [CrossRef] [PubMed]
29. Glass, G.; Papin, J.A.; Mandell, J.W. SIMPLE: A sequential immunoperoxidase labeling and erasing method.
J. Histochem. Cytochem. 2009, 57, 899–905. [CrossRef] [PubMed]
30. Zhang, W.; Hubbard, A.; Jones, T.; Racolta, A.; Bhaumik, S.; Cummins, N.; Zhang, L.; Garsha, K.; Ventura, F.;
Lefever, M.R.; et al. Fully automated 5-plex fluorescent immunohistochemistry with tyramide signal
amplification and same species antibodies. Lab. Investig. 2017, 97, 873–885. [CrossRef] [PubMed]
31. Buchwalow, I.; Samoilova, V.; Boecker, W.; Tiemann, M. Multiple immunolabeling with antibodies from the
same host species in combination with tyramide signal amplification. Acta Histochem. 2018, 120, 405–411.
[CrossRef] [PubMed]
Cancers 2019, 11, 283 18 of 22
32. Prichard, J.W. Overview of automated immunohistochemistry. Arch. Pathol. Lab. Med. 2014, 138, 1578–1582.
[CrossRef] [PubMed]
33. Ilie, M.; Beaulande, M.; Ben Hadj, S.; Chamorey, E.; Schiappa, R.; Long-Mira, E.; Lassalle, S.; Butori, C.;
Cohen, C.; Leroy, S.; et al. Chromogenic Multiplex Immunohistochemistry Reveals Modulation of the
Immune Microenvironment Associated with Survival in Elderly Patients with Lung Adenocarcinoma.
Cancers 2018, 10. [CrossRef] [PubMed]
34. Dixon, A.R.; Bathany, C.; Tsuei, M.; White, J.; Barald, K.F.; Takayama, S. Recent developments in multiplexing
techniques for immunohistochemistry. Expert Rev. Mol. Diagn. 2015, 15, 1171–1186. [CrossRef] [PubMed]
35. Chen, T.; Srinivas, C. Group sparsity model for stain unmixing in brightfield multiplex
immunohistochemistry images. Comput. Med. Imaging Graph. 2015, 46, 30–39. [CrossRef] [PubMed]
36. Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.;
Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors
predict clinical outcome. Science 2006, 313, 1960–1964. [CrossRef] [PubMed]
37. Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-infiltrating
FOXP3 + T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009, 27,
186–192. [CrossRef] [PubMed]
38. Badoual, C.; Hans, S.; Rodriguez, J.; Peyrard, S.; Klein, C.; Agueznay Nel, H.; Mosseri, V.; Laccourreye, O.;
Bruneval, P.; Fridman, W.H.; et al. Prognostic value of tumor-infiltrating CD4 + T-cell subpopulations in
head and neck cancers. Clin. Cancer Res. 2006, 12, 465–472. [CrossRef] [PubMed]
39. Granier, C.; Vinatier, E.; Colin, E.; Mandavit, M.; Dariane, C.; Verkarre, V.; Biard, L.; El Zein, R.; Lesaffre, C.;
Galy-Fauroux, I.; et al. Multiplexed Immunofluorescence Analysis and Quantification of Intratumoral PD-1
+ Tim-3 + CD8 + T Cells. J. Vis. Exp. 2018. [CrossRef] [PubMed]
40. Anichini, A.; Tassi, E.; Grazia, G.; Mortarini, R. The non-small cell lung cancer immune landscape:
Emerging complexity, prognostic relevance and prospective significance in the context of immunotherapy.
Cancer Immunol. Immunother. 2018, 67, 1011–1022. [CrossRef] [PubMed]
41. Remark, R.; Becker, C.; Gomez, J.E.; Damotte, D.; Dieu-Nosjean, M.C.; Sautes-Fridman, C.; Fridman, W.H.;
Powell, C.A.; Altorki, N.K.; Merad, M.; et al. The non-small cell lung cancer immune contexture. A major
determinant of tumor characteristics and patient outcome. Am. J. Respir. Crit. Care Med. 2015, 191, 377–390.
[CrossRef] [PubMed]
42. Rakaee, M.; Busund, L.T.; Paulsen, E.E.; Richardsen, E.; Al-Saad, S.; Andersen, S.; Donnem, T.; Bremnes, R.M.;
Kilvaer, T.K. Prognostic effect of intratumoral neutrophils across histological subtypes of non-small cell
lung cancer. Oncotarget 2016, 7, 72184–72196. [CrossRef] [PubMed]
43. Ilie, M.; Hofman, V.; Ortholan, C.; Bonnetaud, C.; Coelle, C.; Mouroux, J.; Hofman, P. Predictive clinical
outcome of the intratumoral CD66b-positive neutrophil-to-CD8-positive T-cell ratio in patients with
resectable nonsmall cell lung cancer. Cancer 2012, 118, 1726–1737. [CrossRef] [PubMed]
44. Donnem, T.; Kilvaer, T.K.; Andersen, S.; Richardsen, E.; Paulsen, E.E.; Hald, S.M.; Al-Saad, S.; Brustugun, O.T.;
Helland, A.; Lund-Iversen, M.; et al. Strategies for clinical implementation of TNM-Immunoscore in resected
nonsmall-cell lung cancer. Ann. Oncol. 2016, 27, 225–232. [CrossRef] [PubMed]
45. Pages, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.;
Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon
cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [CrossRef]
46. Chiari, R.; Sidoni, A.; Metro, G. Early stage resectable non-small cell lung cancer: Is neoadjuvant
immunotherapy the right way forward? J. Thorac. Dis. 2018, 10, S3890–S3894. [CrossRef] [PubMed]
47. Cottrell, T.R.; Thompson, E.D.; Forde, P.M.; Stein, J.E.; Duffield, A.S.; Anagnostou, V.; Rekhtman, N.;
Anders, R.A.; Cuda, J.D.; Illei, P.B.; et al. Pathologic features of response to neoadjuvant anti-PD-1 in
resected non-small-cell lung carcinoma: A proposal for quantitative immune-related pathologic response
criteria (irPRC). Ann. Oncol. 2018, 29, 1853–1860. [CrossRef] [PubMed]
48. Remon, J.; Vilarino, N.; Reguart, N. Immune checkpoint inhibitors in non-small cell lung cancer (NSCLC):
Approaches on special subgroups and unresolved burning questions. Cancer Treat. Rev. 2018, 64, 21–29.
[CrossRef] [PubMed]
49. Zinger, A.; Cho, W.C.; Ben-Yehuda, A. Cancer and Aging—The Inflammatory Connection. Aging Dis. 2017,
8, 611–627. [CrossRef] [PubMed]
Cancers 2019, 11, 283 19 of 22
50. Shah, W.; Yan, X.; Jing, L.; Zhou, Y.; Chen, H.; Wang, Y. A reversed CD4/CD8 ratio of tumor-infiltrating
lymphocytes and a high percentage of CD4 (+) FOXP3 (+) regulatory T cells are significantly associated with
clinical outcome in squamous cell carcinoma of the cervix. Cell Mol. Immunol. 2011, 8, 59–66. [CrossRef]
[PubMed]
51. Chee, S.J.; Lopez, M.; Mellows, T.; Gankande, S.; Moutasim, K.A.; Harris, S.; Clarke, J.; Vijayanand, P.;
Thomas, G.J.; Ottensmeier, C.H. Evaluating the effect of immune cells on the outcome of patients
with mesothelioma. Br. J. Cancer 2017, 117, 1341–1348. [CrossRef] [PubMed]
52. Han, S.; Zhang, C.; Li, Q.; Dong, J.; Liu, Y.; Huang, Y.; Jiang, T.; Wu, A. Tumour-infiltrating CD4 (+) and CD8
(+) lymphocytes as predictors of clinical outcome in glioma. Br. J. Cancer 2014, 110, 2560–2568. [CrossRef]
[PubMed]
53. Ilie, M.; Hofman, V.; Dietel, M.; Soria, J.C.; Hofman, P. Assessment of the PD-L1 status by
immunohistochemistry: Challenges and perspectives for therapeutic strategies in lung cancer patients.
Virchows Arch. 2016, 468, 511–525. [CrossRef] [PubMed]
54. Kayser, G.; Csanadi, A.; Otto, C.; Plones, T.; Bittermann, N.; Rawluk, J.; Passlick, B.; Werner, M. Simultaneous
multi-antibody staining in non-small cell lung cancer strengthens diagnostic accuracy especially in small
tissue samples. PLoS ONE 2013, 8, e56333. [CrossRef] [PubMed]
55. Selves, J.; Long-Mira, E.; Mathieu, M.C.; Rochaix, P.; Ilie, M. Immunohistochemistry for Diagnosis of
Metastatic Carcinomas of Unknown Primary Site. Cancers 2018, 10, 108. [CrossRef] [PubMed]
56. Cruz, A.F.; Parra, E.R.; Jiang, M.; Fujimoto, J.; Chow, C.W.; Rodriguez-Canales, J.; Behrens, C.; Kalhor, N.;
Weissferdt, A.; Heymach, J.; et al. Characterization of the Immunologic Intra-Tumor Heterogeneity in Early
Stages of Non-Small Cell Lung. Cancer by Multiplex Immunofluorescence. In Proceedings of the IASLC
19th World Conference on Lung Cancer, Toronto, ON, Canada, 23–26 September 2018; pp. S325–S326.
57. Schalper, K.A.; Carvajal-Hausdorf, D.; McLaughlin, J.; Altan, M.; Velcheti, V.; Gaule, P.; Sanmamed, M.F.;
Chen, L.; Herbst, R.S.; Rimm, D.L. Differential Expression and Significance of PD-L1, IDO-1, and B7-H4 in
Human Lung Cancer. Clin. Cancer Res. 2017, 23, 370–378. [CrossRef] [PubMed]
58. Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1,
IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8 (+) T cells. Sci. Transl. Med.
2013, 5, 200ra116. [CrossRef] [PubMed]
59. Schalper, K.A.; Brown, J.; Carvajal-Hausdorf, D.; McLaughlin, J.; Velcheti, V.; Syrigos, K.N.; Herbst, R.S.;
Rimm, D.L. Objective measurement and clinical significance of TILs in non-small cell lung cancer. J. Natl.
Cancer Instig. 2015, 107. [CrossRef] [PubMed]
60. Parra, E.R.; Villalobos, P.; Behrens, C.; Jiang, M.; Pataer, A.; Swisher, S.G.; William, W.N., Jr.; Zhang, J.; Lee, J.;
Cascone, T.; et al. Effect of neoadjuvant chemotherapy on the immune microenvironment in non-small
cell lung carcinomas as determined by multiplex immunofluorescence and image analysis approaches.
J. Immunother. Cancer 2018, 6, 48. [CrossRef] [PubMed]
61. Mami-Chouaib, F.; Blanc, C.; Corgnac, S.; Hans, S.; Malenica, I.; Granier, C.; Tihy, I.; Tartour, E.
Resident memory T cells, critical components in tumor immunology. J. Immunother. Cancer 2018, 6, 87.
[CrossRef] [PubMed]
62. Nizard, M.; Roussel, H.; Diniz, M.O.; Karaki, S.; Tran, T.; Voron, T.; Dransart, E.; Sandoval, F.; Riquet, M.;
Rance, B.; et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun.
2017, 8, 15221. [CrossRef] [PubMed]
63. Ganesan, A.P.; Clarke, J.; Wood, O.; Garrido-Martin, E.M.; Chee, S.J.; Mellows, T.; Samaniego-Castruita, D.;
Singh, D.; Seumois, G.; Alzetani, A.; et al. Tissue-resident memory features are linked to the magnitude of
cytotoxic T cell responses in human lung cancer. Nat. Immunol. 2017. [CrossRef] [PubMed]
64. Djenidi, F.; Adam, J.; Goubar, A.; Durgeau, A.; Meurice, G.; de Montpreville, V.; Validire, P.; Besse, B.;
Mami-Chouaib, F. CD8 + CD103 + tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory
T cells and a prognostic factor for survival in lung cancer patients. J. Immunol. 2015, 194, 3475–3486.
[CrossRef] [PubMed]
65. Mezheyeuski, A.; Bergsland, C.H.; Backman, M.; Djureinovic, D.; Sjoblom, T.; Bruun, J.; Micke, P.
Multispectral imaging for quantitative and compartment-specific immune infiltrates reveals distinct immune
profiles that classify lung cancer patients. J. Pathol. 2018, 244, 421–431. [CrossRef] [PubMed]
Cancers 2019, 11, 283 20 of 22
66. Granier, C.; De Guillebon, E.; Blanc, C.; Roussel, H.; Badoual, C.; Colin, E.; Saldmann, A.; Gey, A.; Oudard, S.;
Tartour, E. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open
2017, 2, e000213. [CrossRef] [PubMed]
67. Song, Z.; Yu, X.; Zhang, Y. Altered expression of programmed death-ligand 1 after neo-adjuvant
chemotherapy in patients with lung squamous cell carcinoma. Lung Cancer 2016, 99, 166–171. [CrossRef]
[PubMed]
68. Mesnage, S.J.L.; Auguste, A.; Genestie, C.; Dunant, A.; Pain, E.; Drusch, F.; Gouy, S.; Morice, P.; Bentivegna, E.;
Lhomme, C.; et al. Neoadjuvant chemotherapy (NACT) increases immune infiltration and programmed
death-ligand 1 (PD-L1) expression in epithelial ovarian cancer (EOC). Ann. Oncol. 2017, 28, 651–657.
[CrossRef] [PubMed]
69. Roussel, H.; De Guillebon, H.; Biard, L.; Mandavit, M.; Gibault, L.; Fabre, E.; Antoine, M.; Hofman, P.;
Beau-Faller, M.; Blons, H.; et al. Composite biomarkers defined by multiparametric immunofluorescence
analysis identify ALK-positive adenocarcinoma as a potential target for immunotherapy. Oncoimmunology
2017, 6, e1286437. [CrossRef] [PubMed]
70. Remon, J.; Hendriks, L.E.; Cabrera, C.; Reguart, N.; Besse, B. Immunotherapy for oncogenic-driven advanced
non-small cell lung cancers: Is the time ripe for a change? Cancer Treat. Rev. 2018, 71, 47–58. [CrossRef]
[PubMed]
71. Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.;
Schultz, K.R.; et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to
PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clin. Cancer Res. 2016,
22, 4585–4593. [CrossRef] [PubMed]
72. Liu, S.Y.; Dong, Z.Y.; Wu, S.P.; Xie, Z.; Yan, L.X.; Li, Y.F.; Yan, H.H.; Su, J.; Yang, J.J.; Zhou, Q.; et al.
Clinical relevance of PD-L1 expression and CD8 + T cells infiltration in patients with EGFR-mutated and
ALK-rearranged lung cancer. Lung Cancer 2018, 125, 86–92. [CrossRef] [PubMed]
73. Badoual, C.; Hans, S.; Merillon, N.; Van Ryswick, C.; Ravel, P.; Benhamouda, N.; Levionnois, E.; Nizard, M.;
Si-Mohamed, A.; Besnier, N.; et al. PD-1-expressing tumor-infiltrating T cells are a favorable prognostic
biomarker in HPV-associated head and neck cancer. Cancer Res. 2013, 73, 128–138. [CrossRef] [PubMed]
74. Granier, C.; Dariane, C.; Combe, P.; Verkarre, V.; Urien, S.; Badoual, C.; Roussel, H.; Mandavit, M.; Ravel, P.;
Sibony, M.; et al. Tim-3 Expression on Tumor-Infiltrating PD-1 + CD8 + T Cells Correlates with Poor Clinical
Outcome in Renal Cell Carcinoma. Cancer Res. 2017, 77, 1075–1082. [CrossRef] [PubMed]
75. Gettinger, S.N.; Choi, J.; Mani, N.; Sanmamed, M.F.; Datar, I.; Sowell, R.; Du, V.Y.; Kaftan, E.; Goldberg, S.;
Dong, W.; et al. A dormant TIL phenotype defines non-small cell lung carcinomas sensitive to immune
checkpoint blockers. Nat. Commun. 2018, 9, 3196. [CrossRef] [PubMed]
76. Parra, E.R.; Behrens, C.; Rodriguez-Canales, J.; Lin, H.; Mino, B.; Blando, J.; Zhang, J.; Gibbons, D.L.;
Heymach, J.V.; Sepesi, B.; et al. Image Analysis-based Assessment of PD-L1 and Tumor-Associated
Immune Cells Density Supports Distinct Intratumoral Microenvironment Groups in Non-small Cell Lung
Carcinom Patients. Clin. Cancer Res. 2016, 22, 6278–6289. [CrossRef] [PubMed]
77. Seyed Jafari, S.M.; Hunger, R.E. IHC Optical Density Score: A New Practical Method for Quantitative
Immunohistochemistry Image Analysis. Appl. Immunohistochem. Mol. Morphol. 2017, 25, e12–e13. [CrossRef]
[PubMed]
78. Koelzer, V.H.; Sirinukunwattana, K.; Rittscher, J.; Mertz, K.D. Precision immunoprofiling by image analysis
and artificial intelligence. Virchows Arch. 2018. [CrossRef] [PubMed]
79. Ruifrok, A.C.; Johnston, D.A. Quantification of histochemical staining by color deconvolution. Anal. Quant.
Cytol. Histol. 2001, 23, 291–299. [PubMed]
80. Thommen, D.S.; Koelzer, V.H.; Herzig, P.; Roller, A.; Trefny, M.; Dimeloe, S.; Kiialainen, A.; Hanhart, J.;
Schill, C.; Hess, C.; et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with
predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 2018, 24, 994–1004.
[CrossRef] [PubMed]
81. Huang, W.; Hennrick, K.; Drew, S. A colorful future of quantitative pathology: Validation of Vectra technology
using chromogenic multiplexed immunohistochemistry and prostate tissue microarrays. Hum. Pathol. 2013,
44, 29–38. [CrossRef] [PubMed]
Cancers 2019, 11, 283 21 of 22
82. Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.;
Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration
and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109.e1110–1119.e1110. [CrossRef] [PubMed]
83. Steele, K.E.; Tan, T.H.; Korn, R.; Dacosta, K.; Brown, C.; Kuziora, M.; Zimmermann, J.; Laffin, B.; Widmaier, M.;
Rognoni, L.; et al. Measuring multiple parameters of CD8+ tumor-infiltrating lymphocytes in human cancers
by image analysis. J. Immunother. Cancer 2018, 6, 20. [CrossRef] [PubMed]
84. Rojo, M.G.; Bueno, G.; Slodkowska, J. Review of imaging solutions for integrated quantitative
immunohistochemistry in the Pathology daily practice. Folia Histochem. Cytobiol. 2009, 47, 349–354.
[CrossRef] [PubMed]
85. Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.;
Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance.
Nature 2014, 515, 568–571. [CrossRef] [PubMed]
86. Sirinukunwattana, K.; Snead, D.R.; Rajpoot, N.M. A Stochastic Polygons Model for Glandular Structures in
Colon Histology Images. IEEE Trans. Med. Imaging 2015, 34, 2366–2378. [CrossRef] [PubMed]
87. Litjens, G.; Kooi, T.; Bejnordi, B.E.; Setio, A.A.A.; Ciompi, F.; Ghafoorian, M.; van der Laak, J.;
van Ginneken, B.; Sanchez, C.I. A survey on deep learning in medical image analysis. Med. Image Anal. 2017,
42, 60–88. [CrossRef] [PubMed]
88. Pantanowitz, L.; Sinard, J.H.; Henricks, W.H.; Fatheree, L.A.; Carter, A.B.; Contis, L.; Beckwith, B.A.;
Evans, A.J.; Lal, A.; Parwani, A.V. Validating whole slide imaging for diagnostic purposes in pathology:
Guideline from the College of American Pathologists Pathology and Laboratory Quality Center. Arch. Pathol.
Lab. Med. 2013, 137, 1710–1722. [CrossRef] [PubMed]
89. Decalf, J.; Albert, M.L.; Ziai, J. New tools for pathology: A user’s review of a highly multiplexed method for
in situ analysis of protein and RNA expression in tissue. J. Pathol. 2018. [CrossRef] [PubMed]
90. Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z.
Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of
Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [CrossRef] [PubMed]
91. Rimm, D.L. Next-gen immunohistochemistry. Nat. Methods 2014, 11, 381–383. [CrossRef] [PubMed]
92. Magaki, S.; Hojat, S.A.; Wei, B.; So, A.; Yong, W.H. An Introduction to the Performance
of Immunohistochemistry. Methods Mol. Biol. 2019, 1897, 289–298. [CrossRef] [PubMed]
93. Torlakovic, E.E.; Cheung, C.C.; D’Arrigo, C.; Dietel, M.; Francis, G.D.; Gilks, C.B.; Hall, J.A.; Hornick, J.L.;
Ibrahim, M.; Marchetti, A.; et al. Evolution of Quality Assurance for Clinical Immunohistochemistry in the
Era of Precision Medicine. Part 3: Technical Validation of Immunohistochemistry (IHC) Assays in Clinical
IHC Laboratories. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 151–159. [CrossRef] [PubMed]
94. Lin, C.Y.; Carneal, E.E.; Lichtensztajn, D.Y.; Gomez, S.L.; Clarke, C.A.; Jensen, K.C.; Kurian, A.W.; Allison, K.H.
Regional Variability in Percentage of Breast Cancers Reported as Positive for HER2 in California: Implications
of Patient Demographics on Laboratory Benchmarks. Am. J. Clin. Pathol. 2017, 148, 199–207. [CrossRef]
[PubMed]
95. Ilie, M.; Long-Mira, E.; Bence, C.; Butori, C.; Lassalle, S.; Bouhlel, L.; Fazzalari, L.; Zahaf, K.; Lalvee, S.;
Washetine, K.; et al. Comparative study of the PD-L1 status between surgically resected specimens
and matched biopsies of NSCLC patients reveal major discordances: A potential issue for anti-PD-L1
therapeutic strategies. Ann. Oncol. 2016, 27, 147–153. [CrossRef] [PubMed]
96. Barnes, M.; Srinivas, C.; Bai, I.; Frederick, J.; Liu, W.; Sarkar, A.; Wang, X.; Nie, Y.; Portier, B.; Kapadia, M.; et al.
Whole tumor section quantitative image analysis maximizes between-pathologists’ reproducibility for clinical
immunohistochemistry-based biomarkers. Lab. Investig. 2017, 97, 1508–1515. [CrossRef] [PubMed]
97. Christgen, M.; von Ahsen, S.; Christgen, H.; Langer, F.; Kreipe, H. The region-of-interest size impacts on
Ki67 quantification by computer-assisted image analysis in breast cancer. Hum. Pathol. 2015, 46, 1341–1349.
[CrossRef] [PubMed]
98. Klauschen, F.; Muller, K.R.; Binder, A.; Bockmayr, M.; Hagele, M.; Seegerer, P.; Wienert, S.; Pruneri, G.;
de Maria, S.; Badve, S.; et al. Scoring of tumor-infiltrating lymphocytes: From visual estimation to
machine learning. Semin. Cancer Biol. 2018, 52, 151–157. [CrossRef] [PubMed]
99. Carey, C.D.; Gusenleitner, D.; Lipschitz, M.; Roemer, M.G.M.; Stack, E.C.; Gjini, E.; Hu, X.; Redd, R.;
Freeman, G.J.; Neuberg, D.; et al. Topological analysis reveals a PD-L1-associated microenvironmental niche
for Reed-Sternberg cells in Hodgkin lymphoma. Blood 2017, 130, 2420–2430. [CrossRef] [PubMed]
Cancers 2019, 11, 283 22 of 22
100. Johnson, D.B.; Bordeaux, J.; Kim, J.Y.; Vaupel, C.; Rimm, D.L.; Ho, T.H.; Joseph, R.W.; Daud, A.I.; Conry, R.M.;
Gaughan, E.M.; et al. Quantitative Spatial Profiling of PD-1/PD-L1 Interaction and HLA-DR/IDO-1 Predicts
Improved Outcomes of Anti-PD-1 Therapies in Metastatic Melanoma. Clin. Cancer Res. 2018, 24, 5250–5260.
[CrossRef] [PubMed]
101. Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schuffler, P.J.; Grolimund, D.;
Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by
mass cytometry. Nat. Methods 2014, 11, 417–422. [CrossRef] [PubMed]
102. Angelo, M.; Bendall, S.C.; Finck, R.; Hale, M.B.; Hitzman, C.; Borowsky, A.D.; Levenson, R.M.; Lowe, J.B.;
Liu, S.D.; Zhao, S.; et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med. 2014, 20, 436–442.
[CrossRef] [PubMed]
103. Nir, G.; Farabella, I.; Perez Estrada, C.; Ebeling, C.G.; Beliveau, B.J.; Sasaki, H.M.; Lee, S.D.; Nguyen, S.C.;
McCole, R.B.; Chattoraj, S.; et al. Walking along chromosomes with super-resolution imaging, contact maps,
and integrative modeling. PLoS Genet. 2018, 14, e1007872. [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access
article distributed under the terms and conditions of the Creative Commons Attribution
(CC BY) license (http://creativecommons.org/licenses/by/4.0/).